
The prevalence of obesity has reached alarming proportions worldwide, and obese individuals are at greater risk for developing chronic kidney disease (CKD) than normal-weight individuals. Although obesity is frequently associated with comorbidities, such as hypertension and diabetes, which are known to contribute to the development of CKD, recent evidence indicates that obesity is a major independent risk factor for renal disease. Obesity-related glomerulopathy has emerged as a distinct clinical entity and its incidence has increased exponentially. However, it is unclear how obesity may lead to CKD in the absence of significant increases in blood pressure (BP).
The Zucker rat (fa/fa) (OZR) is a model of obesity that develops early in life, when increases in BP are not significant. Our preliminary data show that OZR develop glomerular injury, as evidenced by proteinuria and albuminuria, despite similar BP levels as compared to their lean counterparts (LZR). Although previous studies have documented glomerular injury in OZR the mechanisms involved in the development and progression of renal disease in OZR remain unclear.
Glomerular hyperfiltration and hyperperfusion, due to lack of adequate renal autoregulation (AR), play key roles in mediating glomerular injury. Accordingly, we found in preliminary studies that OZR respond with larger increases in glomerular filtration rate (GFR) than LZR following stepwise increases in renal perfusion pressure, indicating an impairment of AR. Modulation of the resistance of preglomerular vasculature is the mechanism underlying AR, based on the myogenic (MYO) response and the tubuloglomerular feedback (TGF) mechanism. Using analysis of dynamic autoregulation of renal blood flow (RBF), we found that one key contributors to efficient AR, the renal MYO response, is impaired in OZR. Therefore, we propose that in obesity there is impaired renal autoregulation that promotes increased transmission of systemic pressure to the glomerulus and subsequent renal injury.
Since BP normally fluctuates during daily activity, inadequate renal AR would lead to transient increases in BP being transmitted to the glomerulus. Baroreceptor reflexes act to rapidly buffer normal fluctuations in BP through adaptive inhibition of sympathetic and stimulation of parasympathetic outflows, respectively. Human obesity is associated with impaired autonomic cardiovascular responses, which are mimicked by the OZR as our preliminary data indicate: compared to LZR they have diminished sensitivity of the baroreflex control of heart rate (BRS) and increased BP variability. In addition, the increased frequency of transient fluctuations of BP into the hypertensive range strongly correlates with the degree of renal injury in OZR. We propose that: in obesity, the kidneys may be exposed to larger and more frequent transient increases in systemic BP, due to inefficient baroreflex buffering of BP variability, which leads to renal injury. However, the mechanisms by which baroreflex function is altered in obesity are not clear.
Along with systemic and renal hemodynamic alterations described above, obesity is also associated with chronic systemic inflammation and increased levels of inflammatory cytokines, such as TNF-a and IL-6. While chronic inflammation is an independent risk factor for cardiovascular disease and CKD, the mechanistic role that inflammation may play in mediating obesity-induced renal injury is not clear. Our previous studies indicate that higher levels of TNF-a and IL-6 in kidneys of OZR increase the density of renal microvasculature with possibly abnormal function. Furthermore, we found that increasing systemic IL-6 in LZR attenuates renal MYO response which may lead to impaired AR. TNF-a and IL-6 also have direct effects on the central nervous system, which may promote autonomic imbalance, and our preliminary data show that IL-6 administration to LZR leads to increased short-term BP variability. We thus further hypothesize that cytokines, particularly TNF-a and IL-6, promote baroreflex dysfunction and impairment of the renal MYO response and AR in OZR, thereby playing a role in the development of renal injury.
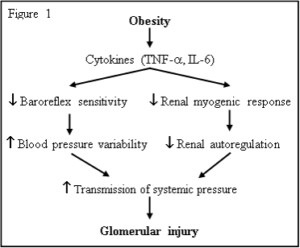
Based on our preliminary data and previously published studies, and as shown in Figure 1, we propose to test the hypothesis that glomerular injury in obesity is due, in part, to impaired renal AR and increased BP variability which result in enhanced transmission of systemic BP load to the glomerulus. We further propose that chronic increases in inflammatory cytokines, TNF-a and/or IL-6, contribute to the attenuation of baroreflex sensitivity and to blunting of the renal MYO response, leading to renal injury in obesity.
We aim to test these hypotheses through an integrative physiological approach, including novel analytical techniques to chronically assess renal AR, blood pressure load and baroreflex function, in conscious, freely moving LZR and OZR. Further, renal microvascular function and structure will be evaluated in pressurized renal interlobar arteries and micro-CT, respectively. Molecular, morphological and biochemical techniques will be used for evaluation of renal glomerular injury.
The proposal has the following specific aims:
Specific Aim 1. Test the hypothesis that impaired renal AR, due to attenuated renal MYO response, and higher BP variability, due to reduced baroreflex sensitivity, cause renal injury in OZR.
Specific Aim 2. Test the hypothesis that increased inflammatory cytokines (specifically, TNF-α and IL-6) cause attenuation of renal MYO response and reduction in baroreflex sensitivity, leading to renal injury in OZR.